
查询时请注意选择相应的产品编号
| 成品预装培养基平皿实用技术手册(一) | |||
|---|---|---|---|
|
微生物污染是药品质量评估的重要指标,为避免外源性污染进入药品生产车间,2010版GMP规定:制药企业需对洁净环境进行动态或静态监测,使用沉降菌平皿、空气浮游菌平皿和表面接触皿评估无菌生产的微生物状况。
过去制药企业一般通过自配培养平皿满足其使用需求,但由于自配培养平皿存在一些难以克服的问题,如:
制备无菌平皿需具备配皿、包装、抽真空、伽马射线终端灭菌等条件,工艺复杂,难以实现,如不进行终端灭菌易造成假阳性结果;
经100%的预培养后,需要对平皿进行无菌性检查,以防带入外来的污染物到监测区;
使用容器装放平皿,或直接将平皿传入洁净区使用,也会污染生产区;
自配的平皿有效期短,常小量多次配制,增加了批间差异和管理成本。
因而,制药企业越来越多的选择使用标准化的商品预灌装培养平皿。随着新版GMP的深入落实,自配平皿将逐渐被预灌装培养平皿取代。
为了加强对洁净区环境监测监督管理,国家食品药品监督局于2008年9月27日下发《关于培养基类产品分类界定的通知》,将培养基类产品列入医疗器械管理,并要求监管部门及生产经营单位遵照执行。培养基类产品分类界定具体内容:即基础、营养、通用增菌、运送、保存培养基等作为Ⅰ类医疗器械管理的培养基产品;而药敏试验用、鉴别、专用/选择增菌、分离、选择培养基作为Ⅱ类医疗器械管理的产品。
2013年11月26日,国家食品药品监督总局在《食品药品监管总局关于印发体外诊断试剂分类子目录的通知》中将培养基类产品划分在《6840 体外诊断试剂分类子目录(2013版)》。
2014年9月5日,国家食品药品监督总局在《关于医疗器械生产质量管理规范执行有关事宜的通告》中第四点指出“自2018年1月1日起,所有医疗器械生产企业应当符合医疗器械生产质量管理规范的要求”。从法规上对培养基类生产作出明确规定。
为帮助广大用户了解和使用商品预灌装培养平皿,我们编写了这本《成品预装培养基平皿实用技术手册》,手册以实践为基础,结合洁净区微生物控制理论,详细的阐明了如何有效控制洁净区微生物才能达到洁净区微生物的稳定、可控制水平,以及洁净环境微生物检测方法及其发展史,阐述在洁净区微生物检测过程中用到的预装成品培养基平皿的生产管理以及质量控制,预装成品培养基平皿的质量要求,该产品是如何使用,在使用过程中常见问题和解答等内容。
本手册共分8章,引入洁净区微生物监测过程的实际案例,也给出如何监测洁净区环境的基本思路,对国外的一些先进理念进行详细的探讨,希望能够帮助大家在使用成品培养基平皿过程得到有效的帮助,使监测结果反映洁净区微生物的真实水平。手册在不同章节中可能出现一些重复的内容,但是这种重复内容是非常有必要的,主要是为了强调其重要性。
本手册除了适用于制药行业外,也适用于医院、疾控中心、食品、以及化妆品、微电子等行业的环境微生物检测。
由于药品、特别是生物制品行业标准一直处于更新状态,加之编者水平有限,时间仓促,手册难免存在不足之处,我们诚心希望各位读者及同行批评指正。
第1章 洁净区微生物控制规范及要求
一、洁净区微生物的控制
药品质量除直接反映在药效和安全性外,还表现在药品质量的稳定性和一致性上,药品生产过程中如果不对生产环境进行严格控制,很容易受到微生物、尘埃粒子等污染或交叉污染。药品的这些特性,对药品生产过程所处的室内环境,对空气的温度、湿度、洁净度、压差、排放方式以及人员卫生的保持等各个方面提出了各种要求。
为了确保药品的质量符合要求,制药企业中药品生产环境需要在洁净区内进行加工完成,洁净区是指将一定空间范围内的空气中的尘埃粒子、微生物等污染物排除,并将室内的温湿度、洁净度、压差、排放方式、振动噪音、照度及静电控制在一定范围内,而给予特别设计的房间。即通过采暖通风和空气调节系统(heating ventilation and air conditioning, HVAC系统)来控制,在中国GMP中称空调净化系统,是制药企业的一个关键系统,它对制药企业能否实现其像患者提供安全有效的药品的目标具有及其重要的影响。如果药品生产环境得到合理的设计、建造、调试、运行和维护保养,则有助于确保产品的质量、提高产品的可靠性,同时降低企业初期的投资成本和后期运行成本。
GMP(2010版)洁净区分类
空气洁净度指洁净环境中空气包含悬浮粒子量的程度,空气中含粒子的数量越少,洁净度越高,世界各国均有相应的洁净度标准,我国的GMP(2010版)规定洁净区的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。
无菌药品生产所需的洁净区可分为以下4个级别:
A级:高风险操作区,如灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。应当有数据证明单向流的状态并经过验证。
在密闭的隔离操作器或手套箱内,可使用较低的风速。
B级:指无菌配制和灌装等高风险操作A级洁净区所处的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作步骤的洁净区。
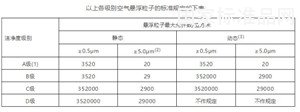
注:
(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。A级洁净区空气悬浮粒子的级别为ISO 4.8,以≥5.0μm的悬浮粒子为限度标准。B级洁净区(静态)的空气悬浮粒子的级别为ISO 5,同时包括表中两种粒径的悬浮粒子。对于C级洁净区(静态和动态)而言,空气悬浮粒子的级别分别为ISO 7和ISO 8。对于D级洁净区(静态)空气悬浮粒子的级别为ISO 8。测试方法可参照ISO14644-1。
(2)在确认级别时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0μm悬浮粒子在远程采样系统的长采样管中沉降。在单向流系统中,应当采用等动力学的取样头。
(3)动态测试可在常规操作、培养基模拟灌装过程中进行,证明达到动态的洁净度级别,但培养基模拟灌装试验要求在“最差状况”下进行动态测试。
|
| 上一篇:适宜的培养基制备方法将影响微生物试验结果 | 下一篇:没有了 |
|---|
无法在这个位置找到: xy/left.htm